Zhi-Yan Zeng1, Jian Chen2, Jarrn-Horng Lin1*
1Department of Materials Science, National University of Tainan, 33, Sec. 2, Shu-lin St., Tainan
2Department of Materials Science and Engineering, Sichuan University of Science and Engineering, Key Lab Material Correction and Protection, Zigong, 643000, People’s Republic of China.
Frontier Research Today, 2018, 1:1007
Published Online: 31 December 2018 (Article)
DOI: 10.31716/frt.201801007
*Corresponding Author. Email: This email address is being protected from spambots. You need JavaScript enabled to view it.
Abstract
One of the challenges for the non-oxidative coupling of methane (NOCM) is to effectively remove the deposited coke over catalysts owing to the over-dehydrogenation of methane. Herein, we show that an in-situ growth of carbon nanotubes (CNTs) instead of coke were observed during NOCM over a CuSO4/γ-Al2O3 catalyst. The as-grown CNTs depict an unexpected promoting effect for NOCM with a highest activity of 0.48 mol kg cat-1·h-1, and maintained 85% activity after 200 h running time. The equilibrium methane conversion is 9.8% with a selectivity of 78.2 % for C2 (C2H4 + C2H6) products. Highly dispersed Cu nanoparticles distributed on the top of CNTs measured by transmission electron microscopy is proposed to result in high catalyst stability during NOCM for 200h instead of deactivation in several hours. Here, we firstly prove that the as-grown CNTs can promote the catalytic activity of NOCM instead of deactivation by coking over catalysts.
Keywords
Carbon nanotube; Non-oxidative coupling of methane; Copper catalysts; Promoting effect; Catalytic activity
Highlight
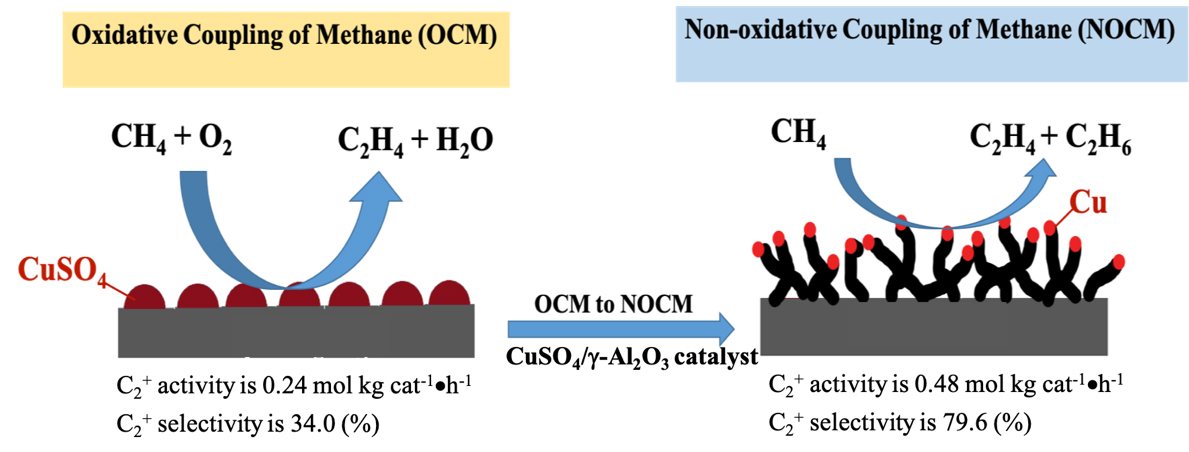
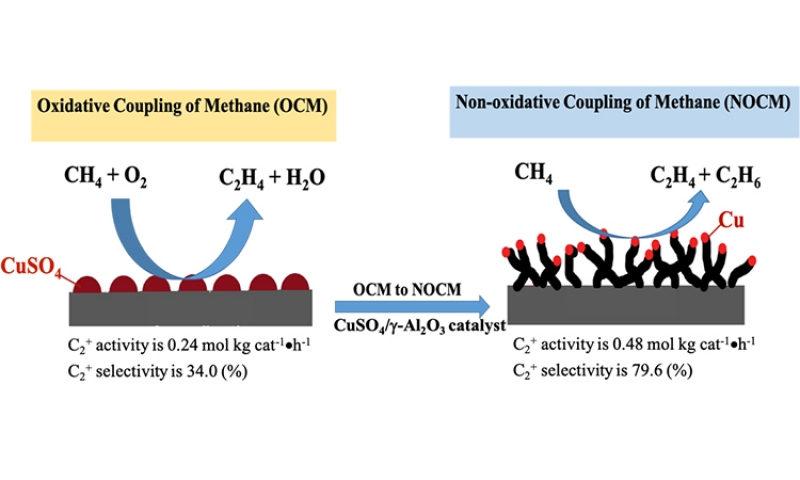
We develop an unique catalyst-5.0 wt% CuSO4/γ-Al2O3 which can conduct both OCM and NOCM. The maximum activity of NOCM is near twice of that for OCM. The as-grown MWCNTs in NOCM is firstly reported to be unexpected promoter over 5.0 wt% CuSO4/γ-Al2O3 catalyst. The yield of the activity is 0.48 mol kgcat-1 hour-1 with a C2 selectivity of 78.2% and an equilibrium methane conversion of 9.8% at 800 oC. Moreover, a 200-h catalytic testing, the activity can maintain at 85% of the highest value. We develop an unique catalyst-5.0 wt% CuSO4/γ-Al2O3 which can conduct both OCM and NOCM. The maximum activity of NOCM is near twice of that for OCM. The as-grown MWCNTs in NOCM is firstly reported to be unexpected promoter over 5.0 wt% CuSO4/γ-Al2O3 catalyst. The yield of the activity is 0.48 mol kgcat-1 hour-1 with a C2 selectivity of 78.2% and an equilibrium methane conversion of 9.8% at 800 oC. Moreover, a 200-h catalytic testing, the activity can maintain at 85% of the highest value.
Introduction
The direct, non-oxidative conversion of methane (NOCM) into light olefins or aromatics, e.g. ethylene, ethane and benzene, is a highly attractive issue for academia and industry. Recently, several advancements have been reported to develop new concepts of catalyst systems for effectively direct converting methane into aromatics or ethylene with remarkable activities, selectivity, and durability1,2. Moreover, the cost of the production of ethylene using NOCM was reported less than one fifth of that by using the stream cracking of crude oil3. Additionally, the main merits of NOCM can get rid of the complicated separation of products without oxygen as well as the generation of CO2. These evidences show that direct conversion of methane is promisingly more economical and environmentally friendly. However, two main challenges for NOCM are needed to be overcome: (i) the activation of methane (C-H bond strength is 434 kJ/mol) need high temperatures (>700 oC), and (ii) catalysts were deactivated quickly through kinetically preferred generation of coke. Therefore, numerous studies conducted the oxidative coupling of methane (OCM) from 1980s4-8. Usually, the presence of oxygen results in the overoxidation of methane, leading to an immense amount of the thermodynamically stable products carbon dioxide and water. Obviously, the carbon utilization efficiency of OCM is relatively low. Thus, a practical route for OCM is not available so far. Listed reactions (1)-(4) of OCM can be represented as
2CH4 (g) + 1/2 O2 (g) → C2H6 (g) + H2O (g) ∆Ho298K = -177 kJmol-1 (1)
C2H6 (g) + 1/2 O2 (g) → C2H4 (g) + H2O (g) ∆Ho298K = -105 kJmol-1 (2)
CH4 (g) + 3/2 O2 (g) → CO (g) + 2H2O (g) ∆Ho298K = -136 kJmol-1 (3)
CH4 (g) + 2 O2 (g) → CO2 (g) + 2H2O (g) ∆Ho298K = -189 kJmol-1 (4)
Recently, Guo et al. demonstrated a new type of heterogeneous iron catalyst which can directly convert methane (48.1% conversion) into higher hydrocarbons (> 99% with ethylene, benzene, and naphthalene) without the formation of coke or unwanted carbon dioxide1. However, preparation of the catalyst was complicated and needed high temperature (1700 oC). More recent, Morejudo et al. used a co-ionic membrane reactor to directly transform methane into benzene with a high carbon efficiency of ~80% at a relative low temperature of 700 oC2. Moreover, several reports have demonstrated that NOCM is a promising route to form light olefins or aromatics9-15. The main reactions of NOCM are described as
CH4 (g) → 1/2 C2H6 (g) + 1/2 H2 (g) ∆Ho298K = 65 kJmol-1 (5)CH4 (g) → 1/2 C2H4 (g) + H2 (g) ∆Ho298K = 101 kJmol-1 (6)
C2H6 (g) → C2H4 (g) + H2 (g) ∆Ho298K = 135 kJmol-1 (7)
Comparison of OCM, the main merit of NOCM is a suppression of over-oxidation of methane. This will significantly improve the selectivity of higher hydrocarbons instead of carbon dioxide. However, how to control the catalysis system preventing over-dehydrogenation of methane is a key step to avoid the formation of coke in NOCM1, 2,9-15. Therefore, the design concepts for active catalysts or reaction systems should avoid completely dehydrogenation of methane in NOCM. Conventionally, the as-generated carbon materials during NOCM were reported that will led to a major deactivation in catalytic activity. The active metal components of catalysts were covered with the as-grown carbon materials with highly graphitic structures, e.g. coke, graphite or carbon nanofibers, and forfeited their catalytic activity gradually1,2,9-15. The catalytic activities of catalysts for NOCM will deactivate soon, normally in less several hours. Therefore, the design concepts of the conventional catalysts were focused on preventing the formation of carbonaceous materials during the reaction conditions in NOCM. As we know, the promotion effect of the as-grown carbonaceous materials in NOCM was not reported yet. Herein, we present a new finding that the as-grown carbon nanotubes (CNTs) during NOCM over CuSO4/γ-Al2O3 catalysts can promote the catalytic activity with high C2 (C2H4 + C2H6) yields for a 200 h test. The in-situ multi-walled CNTs growth following a tip-growth model through Cu nanoparticles (NPs) during the NOCM reaction was observed. The highly-dispersed Cu NPs on the top of the as-grown MWCNTs conducted an admirable activity of NOCM with a remarkable resistance of deactivation. These results present new concepts for NOCM.
Methods
Materials
Copper (II) sulfate pentahydrate (99.0%) was purchased from Signa-Aldrich. γ-Al2O3 powder was supplied by Degussa co. They were used as received without further purification. Methane, oxygen, air, and argon gas were purchased from Air Liquide.
Preparation of copper catalysts
Copper catalysts were prepared by wet impregnation according to our earlier work16, 17. Typically, copper (II) sulfate pentahydrate (0.1 g) was dissolved in 10 mL pure water with a vigorous magnetic stirring under air. The copper solution was added into γ-Al2O3 powder (0.4 g) step by step with a potent stirring by hand for 30 min. The final slurry solution was dried in oven at 120 oC under air overnight. The final loading of Cu was 5.0 ± 0.2 wt. %, which was determined by atomic adsorption spectroscopy and inductively coupled plasma mass spectrometry (ICP-MS, PE-SCIEX ELAN 6100 DRC).
Oxidative conversion of methane (OCM)
Catalytic activities were carried out in a continuous flow, fixed-bed quartz tube reactor. In all OCM tests, 0.1 g of the prepared copper catalysts-5.0 wt% CuSO4/γ-Al2O3 was put in the reactor. The reaction temperatures were adjusted between 700-1100 oC. The feed gas is a mixture of argon diluted methane (CH4/O2/Ar = 60/1/19) with a flow rate of 40 mL/min. The effluent gas composition was examined by an online gas chromatography (GC, Shimadzu GC-2014), which is equipped with an FID detector with HP-DPX5 column (I.D. is 0.53 mm, 25 m in length with a 1.0 mm inner coating film). Methane conversion, hydrocarbon products selectivity and carbon deposition were calculated through the carbon balance, following previously reported methods13-15.
Non-oxidative conversion of methane (NOCM)
Catalytic activities were carried out in a continuous flow, fixed-bed quartz tube reactor. In all NOCM tests, 0.1 g of the prepared copper catalysts-5 wt% CuSO4/γ-Al2O3 was put in the reactor. The reaction temperatures were adjusted between 700-1100 oC. The feed gas is a mixture of argon diluted methane (Ar/CH4 = 1/3) with a flow rate of 40 mL/min. The effluent gas composition analysis and characterization of methane conversion, product distribution were carried out the same standards of OCM.
Catalyst characterization
Scanning electron microscopy (SEM) analysis was conducted using a JEOL JSM-6700F. Transmission electron microscopy (TEM, JEOL AEM-300 and JEM-2100) equipped with an energy dispersive spectrometer (EDS) were used to investigate the micro- and nano-scale structural morphologies of the as-grown samples and perform elemental analyses. High resolution TEM Images were performed on to investigate the micro- and nano-scale structural morphologies of Cu NPs. The weight percent of the as-grown carbon materials was analyzed by a thermogravimeter analyzer (TGA, TA-Q500). The oxidative characteristics of the samples were performed in TGA under air atmosphere (40 mL/min) with a ramp of 40 oC/min during 30-800 oC. X-ray diffraction data analysis was performed by a Miniflex-III (Rigaku) with a Cu Kα radiation source (λ=0.15418 nm). Raman scattering spectroscopy (JOBIN-YVON T64000) with a laser excitation wavelength of 532 nm was used to characterize the graphite-amorphous carbon features of the as-grown carbon materials.
Results and Discussion
Previously, sulfate-assisted metal catalysts have been reported with high activities in OCM18-20. The main concepts of these catalysts were regarded that the sulfated-supports, e.g. SO4-2-MgO or SO4-2-ZrO2, can modify surface acidity of catalysts then conduct higher dehydrogenation rate of methane. In OCM, the present of oxygen can prevent over-dehydrogenation of methane and recover active sites of catalysts through the redox process. However, this is a trade-off in OCM and usually leads to a difficult separation of gas products. Therefore, the employment of OCM in practical utilization has been postponed for several decades. Originally, we employ CuSO4/γ-Al2O3 as a catalyst according to several previous reports for studying OCM. However, we accidentally found that CuSO4/γ-Al2O3 catalyst displayed remarkable activities in growth of MWCNTs with chemical vapor deposition with methane21, ethylene22 and ethanol23. These reliable evidences lead us to conduct both OCM and NOCM over CuSO4/γ-Al2O3 catalyst for an adequate reaction time.
To check the activity of the copper sulfated catalyst in the dehydrogenation of methane with and without oxygen, we conducted the activities of OCM firstly and then followed NOCM over 5.0 wt% CuSO4/γ-Al2O3 catalyst at 800 oC. In the left-upper panel of Figure 1, which demonstrates that the reactivity of 5.0 wt% CuSO4/γ-Al2O3 in OCM was stable and maintained approximately at the space-time yield of 0.24 mol kgcat-1 hour-1 for 2 h. After OCM, oxygen was replaced by argon, then switched to the NOCM mode. It is clearly to find that the reactivity was sharply dropped to zero in 30 min. However, surprisingly, the reactivity can recover gradually in 180 min of the NOCM process, and reached the maximum of reactivity with a yield of 0.48 mol kgcat-1 hour-1, which is twice of the value of OCM. Moreover, in NOCM process, the first 90 minutes (reaction time during 150-240 minutes in Figure 1), the reactivity presents a lower slope (r1), and the later 60 minutes (reaction time during 240-300 minutes) demonstrates a higher slope (r2) of reactivity. Apparently, the slope of r2 is higher than r1. This indicates that the recovery behaviors of reactivity over 5.0 wt% CuSO4/γ-Al2O3 during NOCM process depict two stages. The first is a slow-growing stage and follows a fast-growing route to reach the maximum of reactivity. We took samples from the reaction times at 30 (a), 60 (b), 120 (c), and 180 (d) min, respectively and examined the surface morphologies of them using SEM, shown in Figure 1(a-d). It is plainly to note that the recovery of catalytic activities over 5.0 wt% CuSO4/γ-Al2O3 catalysts is accompanying with the growth of as-grown CNTs or CNFs. The life-time testing was performed for 200 h at 800 oC, the 5.0 wt% CuSO4/γ-Al2O3 was quite stable, and displayed only a slight decline in reaction activity, shown in upper-right panel of Figure 1.
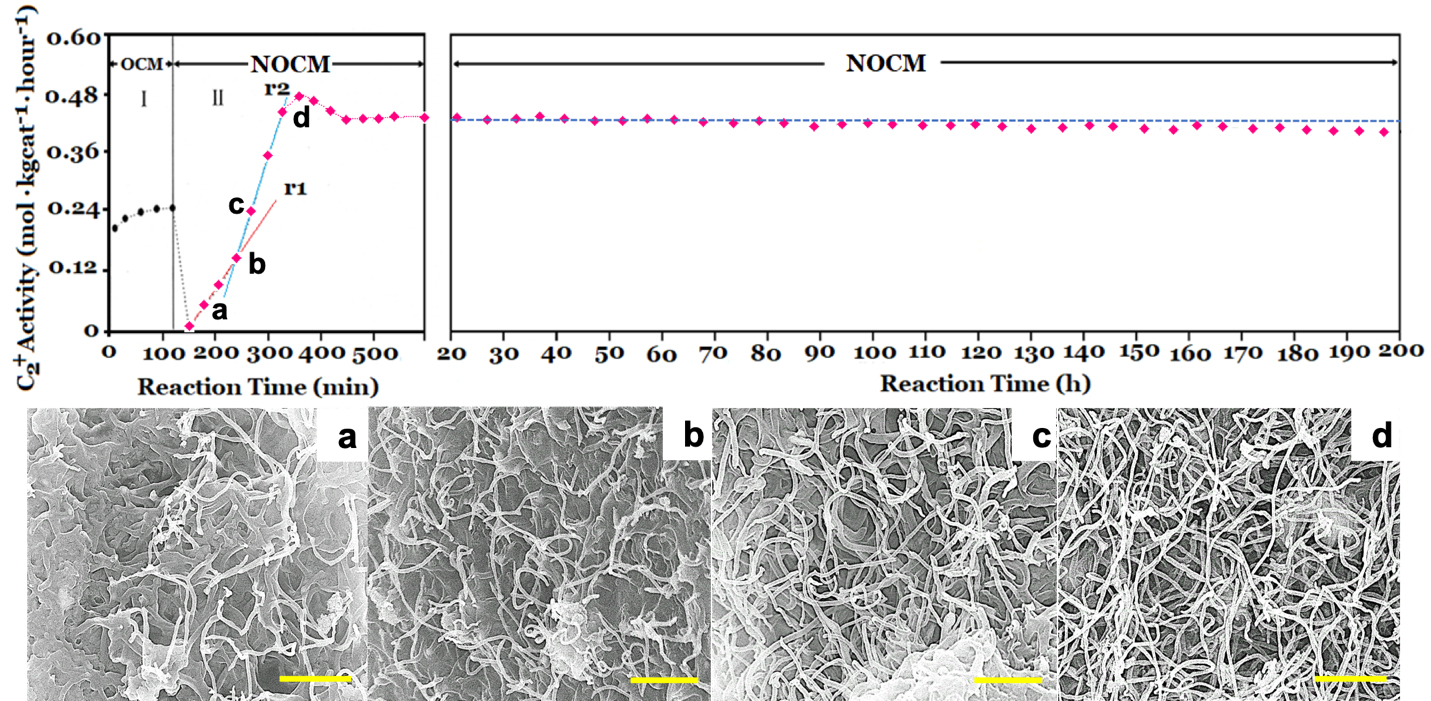
Figure 1. The left-upper and right-upper panels: Catalytic activity of 5.0 wt% CuSO4/γ-Al2O3 catalyst for oxidative coupling of methane (OCM) and non-oxidative coupling of methane (NOCM) at 800 oC. For OCM, the feeding gas is a mixture of CH4/O2/Ar = 60/1/19 with a flow rate of 40 mL/min. For NOCM, the feeding gas is a mixture of CH4/Ar = 3/1 with a flow rate of 40 mL/min. (a-d) are SEM images of the different reaction time position in the upper-left panel a-d for NOCM. The scale bar in (a-d) is 0.5 mm.
For understanding the morphologies of as-grown fiber-like structures during NOCM over 5.0 wt% CuSO4/γ-Al2O3 catalyst at 800 oC, we examined the sample using TEM and selected-area electron diffraction (SAED). Figure 2a displays that Cu NPs on the top position of MWCNTs are the major product and amorphous carbon (a-C) is formed anywhere on the side-wall of MWCNTs. Figure 2b is an enlarged area of Figure 2a. The SAED on the top position of a typical MWCNT, shown in Figure 2c, depicts that metallic Cu is the major component of Cu NPs. The results shown in Figure 2a-c reveal that the as-grown MWCNTs were formed through a top-growth model over Cu NPs. Simultaneously, Cu NPs were highly re-dispersed on the top of MWCNTs, which prevents Cu aggregating or deactivation owing to coking. A detailed analysis of the as-grown MWCNTs and amorphous carbon were characterized by TGA and Raman spectra, shown in Figure 3a and 3b, respectively. The TGA oxidation profile of the as-grown MWCNTs and of the amorphous carbon over 5.0 wt% CuSO4/γ-Al2O3 catalyst is displayed in Figure 3a. Two peaks are clearly shown in the first-derivative curve of the TGA profile. Accordingly, the low-temperature peak (446 oC) is assigned to the combustion of amorphous carbon and the higher temperature (515 oC) is assigned to that of MWCNTs16, 17. The growth yield of the as-grown carbon soot is approximately 30.4 wt% with a composition of approximately 44 % amorphous carbon and 56 % MWCNTs. The graphitic quality of the as-grown MWCNTs was determined using the intensity ration of the G-band (tangential mode of graphite~1343 cm-1) and D-band (defect mode~1600 cm-1). Figure 3b displays that the IG/ID (~1.0) ratio is a typical feature of MWCNTs. The yields of the as-grown MWCNTs at various reaction times over 5.0 wt% CuSO4/γ-Al2O3 catalyst in NOCM in the initial 150 min was plotted in Figure 3c. A linear relationship between the MWCNT yields and reaction times was observed. Combined with the results in the left-upper panel of Figure 1, it is clearly to note that the growth of MWCNTs is the decisive step in NOCM over 5.0 wt% CuSO4/γ-Al2O3 catalyst. The average particle size of Cu NPs calculated using XRD patterns of Cu (111) crystalline (2θ = 43.3o) with various reaction times in NOCM also illustrated in Figure 3d. A slightly raising size of Cu NPs is not the main factor to dominate the catalytic activity for NOCM.

Figure 2. (a) A typical TEM image of the as-grown CNTs, (b) TEM image for the enlarge area of (a), and (c) a selected-area electron diffraction of Cu NPs of (b), over 5.0 wt% CuSO4/γ-Al2O3 catalyst in NOCM at 800 oC under a mixture of Ar diluted methane with a flow rate of 40 mL/min for 2 hours.
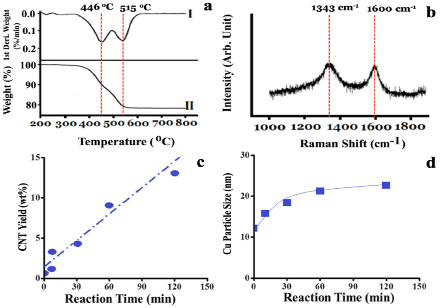
Figure 3. (a) Oxidative TGA profiles and (b) Raman spectra of the samples over 5.0 wt% CuSO4/γ-Al2O3 catalyst in NOCM at 800 oC under a mixture of Ar diluted methane with a flow rate of 40 mL/min for 2 hours. (c) the CNT yield versus the reaction time over 5.0 wt% CuSO4/γ-Al2O3 catalyst in NOCM at 800 oC under a mixture of Ar diluted methane with a flow rate of 40 mL/min. (d) the average particle size of Cu NPs calculated through XRD patterns of Cu (111) at 2θ = 43.3o based on Scherrer equation.
For NOCM, CH4 conversion, product selectivity and life time are three main factors to evaluate the catalytic performance of 5.0 wt% CuSO4/γ-Al2O3 catalyst. In Figure 1, we demonstrate that the life time in NOCM can maintain 200 h with a slight decay of activity. Figure 4a describes the trends of CH4 conversions and C2+ selectivities (C2H4, C2H6, and C4H10, mainly) with various reaction times in NOCM at 800 oC over 5.0 wt% CuSO4/γ-Al2O3 catalyst. Apparently, the initial CH4 conversion is near 29.8 %, however, which descended quickly in 30 min. and maintained at a stable value of 9.8 % after 150 min. testing time. Selectivity to ethylene plus ethane (78.2 %), and butadiene (1.6%) were constant while the reaction time was above 150 minutes. Although activity and conversion of NOCM over 5 wt% CuSO4/γ-Al2O3 is not the highest compared with previous reports1, 2, however, the formation of carbon-related materials didn’t obey the previous concept as a role of deactivation for NOCM. On the contrary, the as-grown carbon-related materials dominated the reaction process in NOCM. By comparison, the pretreatments of 5.0 wt% CuSO4/γ-Al2O3 catalyst were conducted at 800 oC under air, argon diluted hydrogen, or helium for 2 hours, the results are presented in Figure 4b I-III, respectively. Interestingly, under helium or hydrogen treatments, the maximum activities would reach in less 100 minutes, however their highest catalytic rates are still lower than that of air-treated sample. The major difference on these treatments was the formation of Cu nanoparticles (NPs). Calculated by XRD patterns of Cu (111) using Scherrer equation, the particle sizes of Cu NPs by helium and hydrogen pretreatments are 27.2 and 32.5 nm, respectively. This perhaps explains why the catalytic activity in NOCM, air-treated sample is highest, owing to slow formation of Cu NPs under methane. The Cu NPs are easily to aggregate during the reaction temperature and reduced its activity. Furthermore, the aggregation of Cu NPs will retard the formation of carbon nanotubes, therefore lower down the catalytic activity in NOCM. Although the various treatments will cause the different catalytic performances, however if the promoted effect should come from the as-grown MWCNTs. The products of ethylene/ethane ratios would be similar in the stable catalytic reaction, Figure 4c supports our consideration. The catalytically initiated reaction was caused by Cu NPs or CuO, so the ethylene/ethane ratios are varying in the beginning, however, the ratios will get to the similar values at about 3.0 after 200-minute reaction time. For understanding the promoted effect of the as-grown CNTs, we conducted the NOCM on 5.0 wt% CuSO4/γ-Al2O3 with various temperatures (600-1100 oC). The results were displayed in Figure 4d-4e. Interestingly, the maximum yield (16.8 wt%) of CNTs (Figure 4d) was occurred at 800 oC, which also performed the highest activity of NOCM, shown in Figure 4e. The trend of catalytic activity is consistent with that of the deposited amount of CNTs, which depicts that the catalytic performance of 5.0 wt% CuSO4/γ-Al2O3 would be promoted by the as-grown CNTs. The decay of activity while the reaction temperature was higher than 800 oC was examined by XRD patterns, shown in Figure 4f. The possible reason is that the support-Al2O3 will transform from γ to θ form, and then finally become the α form when temperature increased above 800 oC. The crystallinity of α-Al2O3 display higher integrity and also indicates that the surface area of Al2O3will highly reduce from γ to α form. Therefore, the intensely aggregation of Cu NPs will happen, this accelerates the deactivation of catalytic activity in NOCM. This demonstrate a new concept that the in-situ growth of CNTs displays an unexpectedly promoting effect for NOCM over 5.0 wt% CuSO4/γ-Al2O3 catalyst.
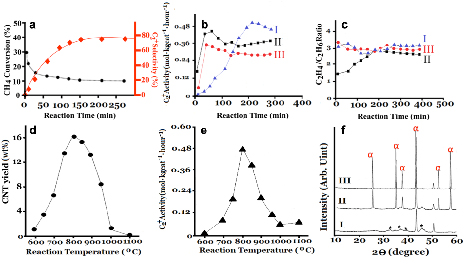
Figure 4. (a) Catalytic performance of 5.0 wt% CuSO4/γ-Al2O3 catalyst for CH4 conversion and C2+ selectivity in NOCM. (b) C2+ activity performance and (c) C2H4/C2H6 yield ratios of (I) Air-treated, (II) H2/N2(=1/3)-treated, and (III) He-treated samples of 5.0 wt% CuSO4/γ-Al2O3 catalyst at 800 oC under a mixture of Ar diluted methane with a flow rate of 40 mL/min. Pretreatment time for (I-III) is 2 hours at 800 oC with a flow rate of 40 mL/min. Profiles of (d) CNT yields and (e) C2+ activities 5.0 wt% CuSO4/γ-Al2O3 catalyst at various reaction temperatures in NOCM under a mixture of Ar diluted methane with a flow rate of 40 mL/min. (f) XRD patterns of 5.0 wt% CuSO4/γ-Al2O3 catalyst at (I) 800, (II)1000, and (III)1100 oC.
Apart from the catalytic performances of 5.0 wt% CuSO4/γ-Al2O3 catalyst for NOCM, it is essential to investigate the active components of 5.0 wt% CuSO4/γ-Al2O3 catalyst. In Figure 2, we note that metallic Cu NPs should be active centers for growing CNTs and promote the transformation of CH4 into light olefins, e.g. C2H6, C2H4…etc., in NOCM. Therefore, we prepared several Cu catalysts using various Cu precursors, such as Cu(CH3COO)2 and Cu(NO3)2, supported on γ-Al2O3 through impregnation. The temperature-programmed reduction profiles of Cu-based catalysts are shown in Figure 5a. Obviously, Cu catalysts prepared by Cu(CH3COO)2 (Figure 5a-I) and Cu(NO3)2 (Figure 5a-II) display reduction peaks at lower temperatures which are assigned to smaller CuO NPs. CuSO4-derived Cu catalyst (Figure 5a-III) conducts similar TPR peak comparing with those of bulk CuO (Figure 5a-V) or air-treated CuO (Figure 5a-IV). However, Cu catalysts derived from Cu(CH3COO)2, Cu(NO3)2, or bulk CuO did not show any catalytic activities in NOCM. Figure 5b-5c demonstrate clear images that Cu catalysts-derived from Cu(CH3COO)2 or Cu(NO3)2 will deactivate quickly. The main reason is that Cu NPs were coated with amorphous carbon through the over dehydrogenation of methane. Interestingly, why CuSO4/γ-Al2O3 catalyst can survive and conduct remarkable catalytic performance in NOCM. The ruling difference is the strong metal-support interaction (SMSI) between CuSO4 and γ-Al2O316, 17, which dominates the growth of CNTs and then promotes the activity in NOCM. Without SMSI, Cu NPs which derived from Cu(CH3COO)2 or Cu(NO3)2 are easy to aggregate and then covered with amorphous carbon. This will cause the deactivation of catalytic activity very soon, usually in several minutes.
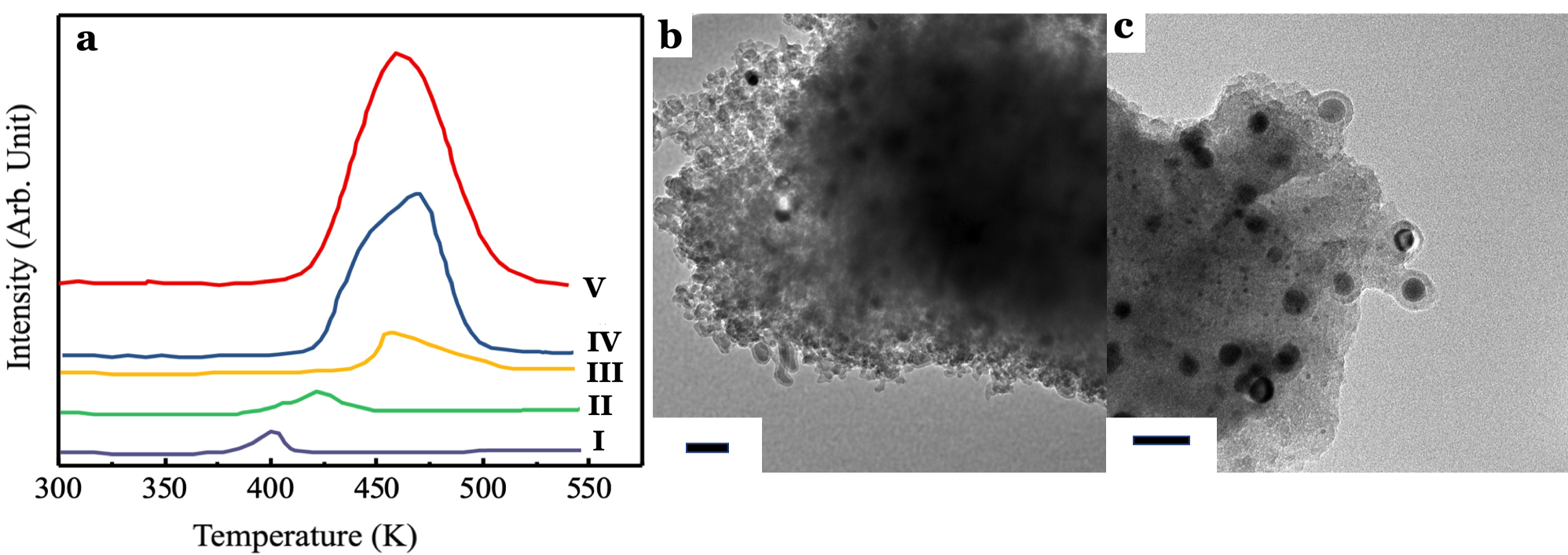
Figure 5. (a) Temperature programmed reduction profiles of 5.0 wt% Cu/γ-Al2O3 catalysts with various Cu precursors (I) Cu(CH3COO)2, (II) Cu(NO3)2, and (III) CuSO4. A comparison TPR profiles of (IV) Air-treated bulk CuO and (V) untreated bulk CuO powder. (b) and (c) are the typical TEM images of samples, 5.0 wt% Cu/γ-Al2O3 catalysts prepared with Cu(CH3COO)2, and Cu(NO3)2, respectively, in NOCM at 800 oC under a mixture of Ar diluted methane with a flow rate of 40 mL/min for 2 hours.
In order to testify our consideration, we examine surface morphologies of CuSO4/γ-Al2O3 catalyst which conducted the NOCM for 120 min, shown in Figure 6. The SEM image demonstrates dense-growth CNTs and CNT-free area on CuSO4/γ-Al2O3 catalyst. Semi-quantitatively elemental analysis of two areas using EDX spectra (spectrum 1 and spectrum 2) are listed. It is obviously to note that CNT-free area presents lower values of S and O elements, which strongly suggests that SOx is the determining component of CuSO4/γ-Al2O3 catalyst.
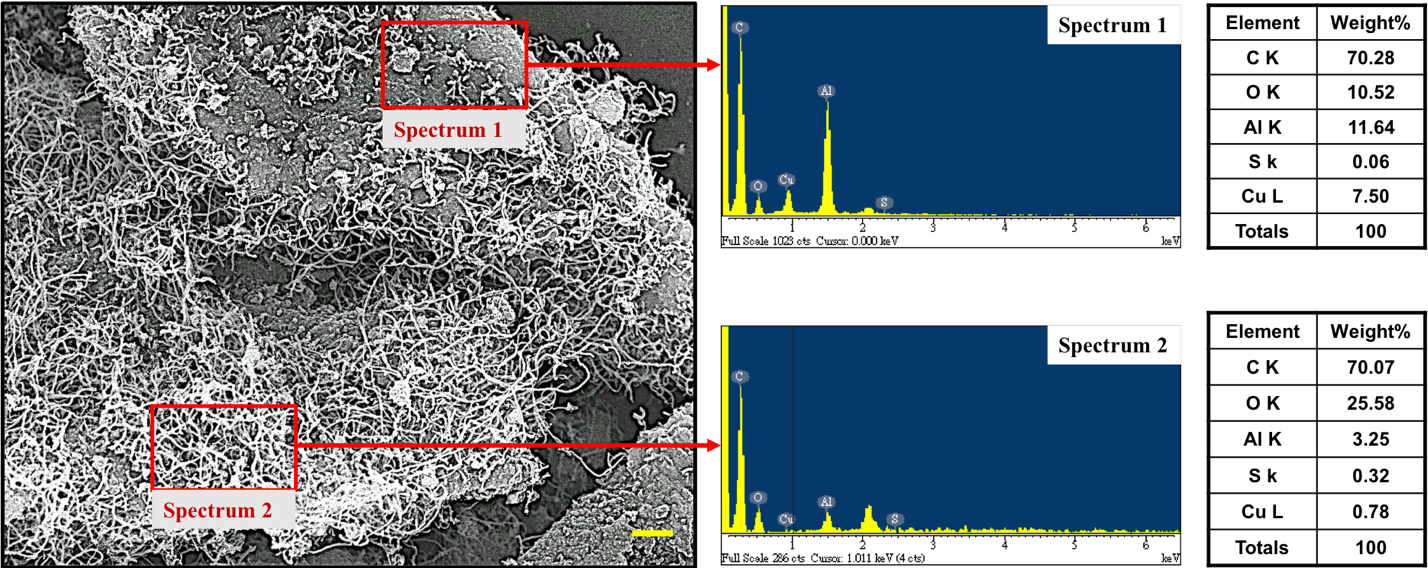
Figure 6. EDS spectra and semi-quantitatively elemental analysis of the two selected area of a typical SEM image of 5.0 wt% CuSO4/γ-Al2O3 catalyst in NOCM at 800 oC under a mixture of Ar diluted methane with a flow rate of 40 mL/min for 2 hours.
Our observations reveal that the switch from OCM to NOCM over 5.0 wt% CuSO4/γ-Al2O3 catalyst will initially generate CNTs instead of conducting the coupling of methane. Therefore, the reactivity of catalyst was quickly dropping to zero, and further instrumentally examined owing to the formation of CNTs. Possibly, this is the main reason why previously studies on NOCM did not disclose the promoting effect of the as-grown carbon materials. In this work, we reveal that the catalytic reactivity will recover gradually in NOCM using 5.0 wt% CuSO4/γ-Al2O3 catalyst with adequate reaction time. As examining the results shown from Figure 1 to Figure 6, we can suppose that the unexpected promoting effect of the as-grown MWCNTs were performed with two stages. MWCNTs were formed in the initial stage of the dehydrogenation of methane through a surface-diffusion mode over copper nanoparticles in 5.0 wt% CuSO4/γ-Al2O3 catalyst. Then Cu NPs were highly dispersed on the top of the tangle-like MWCNTs, which mainly conducted the route of coupling of methane into ethylene, ethane and higher hydrocarbons. This interesting result was not reported previous.
Conclusions
Here, we demonstrate 5.0 wt% CuSO4/γ-Al2O3 catalyst can perform both OCM and NOCM. The maximum activity of NOCM is near twice of that for OCM. The as-grown MWCNTs is firstly reported to be unexpected promoter for 5.0 wt% CuSO4/γ-Al2O3 catalyst in NOCM. The yield of the activity is 0.48 mol kgcat-1 hour-1 with a C2 selectivity of 78.2% and an equilibrium methane conversion of 9.8% at 800 oC. A 200-h catalytic testing, the activity can maintain at 85% of the highest value. We explore a new concept for direct converting of methane with the promotion of the as-grown carbon materials instead of deactivation.
Acknowledgements
This work is supported by the Ministry of Science and Technology of Taiwan. (MOST 104-2113-M-024- and MOST 105-2113-M-024 -001).
Notes
The authors declare no competing financial interest.
References
[1] Guo X, Fang G, Li G, Ma H, Fan H, Yu L, et al. Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen. Science 2014;344:616–9. doi:10.1126/science.1253150
[2] Morejudo SH, Zanón R, Escolástico S, Yuste-Tirados I, Malerød-Fjeld H, Vestre PK, et al. Direct conversion of methane to aromatics in a catalytic co-ionic membrane reactor. Science 2016;353:563–6. doi:10.1126/science.aag0274
[3] Sheng H, Schreiner EP, Zheng W, Lobo RF. Non-oxidative Coupling of Methane to Ethylene Using Mo2 C/[B]ZSM-5. ChemPhysChem 2018;19:504–11. doi:10.1002/cphc.201701001
[4] KELLER G. Synthesis of ethylene via oxidative coupling of methane I. Determination of active catalysts. Journal of Catalysis 1982;73:9–19. doi:10.1016/0021-9517(82)90075-6
[5] OTSUKA K. Active and selective catalysts for the synthesis of C2H4 and C2H6 via oxidative coupling of methane. Journal of Catalysis 1986;100:353–9. doi:10.1016/0021-9517(86)90102-8
[6] Amenomiya Y, Birss VI, Goledzinowski M, Galuszka J, Sanger AR. Conversion of Methane by Oxidative Coupling. Catalysis Reviews 1990;32:163–227. doi:10.1080/01614949009351351
[7] Lunsford JH. The Catalytic Oxidative Coupling of Methane. Angewandte Chemie International Edition in English 1995;34:970–80. doi:10.1002/anie.199509701
[8] Lunsford JH. Catalytic conversion of methane to more useful chemicals and fuels: a challenge for the 21st century. Catalysis Today 2000;63:165–74. doi:10.1016/S0920-5861(00)00456-9
[9] Chen L, Lin L, Xu Z, Zhang T, Li X. Promotional effect of Pt on non-oxidative methane transformation over Mo-HZSM-5 catalyst. Catalysis Letters 1996;39:169–72. doi:10.1007/BF00805578
[10] Soulivong D, Norsic S, Taoufik M, Coperet C, Thivolle-Cazat J, Chakka S, et al. Non-Oxidative Coupling Reaction of Methane to Ethane and Hydrogen Catalyzed by the Silica-Supported Tantalum Hydride: (≡SiO)2 Ta−H. Journal of the American Chemical Society 2008;130:5044–5. doi:10.1021/ja800863x
[11] Lukyanov DB, Vazhnova T. Transformation of methane over platinum supported catalysts at moderate temperature. Journal of Molecular Catalysis A: Chemical 2011;342-343:1–5. doi:10.1016/j.molcata.2011.04.011
[12] Li L, Mu X, Liu W, Kong X, Fan S, Mi Z, et al. Thermal Non-Oxidative Aromatization of Light Alkanes Catalyzed by Gallium Nitride. Angewandte Chemie International Edition 2014;53:14106–9. doi:10.1002/anie.201408754
[13] Nishikawa Y, Ogihara H, Yamanaka I. Liquid-Metal Indium Catalysis for Direct Dehydrogenative Conversion of Methane to Higher Hydrocarbons. ChemistrySelect 2017;2(16):4572–6. http://doi.org/10.1002/slct.201700734
[14] Gerceker D, Motagamwala AH, Rivera-Dones KR, Miller JB, Huber GW, Mavrikakis M, et al. Methane Conversion to Ethylene and Aromatics on PtSn Catalysts. ACS Catalysis 2017;7:2088–100. doi:10.1021/acscatal.6b02724
[15] Xiao Y, Varma A. Highly Selective Nonoxidative Coupling of Methane over Pt-Bi Bimetallic Catalysts. ACS Catalysis 2018;8:2735–40. doi:10.1021/acscatal.8b00156
[16] Lin Y-C, Lin J-H. Purity-controllable growth of bamboo-like multi-walled carbon nanotubes over copper-based catalysts. Catalysis Communications 2013;34:41–4. doi:10.1016/j.catcom.2013.01.013
[17] Lin J-H, Chen C-S, Zeng Z-Y, Chang C-W, Chen H-W. Sulphate-activated growth of bamboo-like carbon nanotubes over copper catalysts. Nanoscale 2012;4:4757. doi:10.1039/c2nr30854a
[18] Murata K, Hayakawa T, Fujita K. Excellent effect of lithium-doped sulfated zirconia catalysts for oxidative coupling of methane to give ethene and ethane. Chemical Communications 1997:221–2. doi:10.1039/a606624k
[19] Kurosaka T, Matsuhashi H, Arata K. Dehydrogenative Coupling of Methane Catalyzed by Platinum-Added Sulfated Zirconia and Characterization of the Catalyst Surface. Journal of Catalysis 1998;179:28–35. doi:10.1007/jcat.1998.2209
[20] Farrell BL, Igenegbai VO, Linic S. A Viewpoint on Direct Methane Conversion to Ethane and Ethylene Using Oxidative Coupling on Solid Catalysts. ACS Catalysis 2016;6:4340–6. doi:10.1021/acscatal.6b01087
[21] Lin J-H, Chen C-S, Ma H-L, Hsu C-Y, Chen H-W. Synthesis of MWCNTs on CuSO4/Al2O3 using chemical vapor deposition from methane. Carbon 2007;45:223–5. doi:10.1016/j.carbon.2006.09.012
[22] Lin J-H, Chen C-S, Rümmeli MH, Zeng Z-Y. Self-assembly formation of multi-walled carbon nanotubes on gold surfaces. Nanoscale 2010;2:2835. doi:10.1039/c0nr00256a
[23] Hsiao C-H, Lin J-H. Growth of a superhydrophobic multi-walled carbon nanotube forest on quartz using flow-vapor-deposited copper catalysts. Carbon 2017;124:637–41. doi:10.1016/j.carbon.2017.09.023
Open Access
This article is licensed under a Creative Commons Attribution 4.0 International License. (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
© The Author(s) 2018